Literature Review
This page presents research efforts conducted in the field of RNA structure prediction.
View the section Overview for background information about RNA molecules.
Problem Statement
The objective of RNA secondary structure prediction is to determine the pairing of nucleotide from their sequence. This can be described as a prediction problem in which:
The input is a sequence of four characters:
A,C,G, andU(i.e. primary structure).The output is s sequence of characters describing the pairing of nucleotides (i.e. secondary structure). This structure can be described in different ways: - The bracket notation uses the characters
.,(, and)todesignate nucleotides that are unpaired, paired with a nucleotide in the 3’ direction, or paired with a nucleotide in the 5’ direction, respectively. It cannot represent pseudo-knots.
The shadow of the secondary structure is a sequence of Boolean values to distinguish paired and unpaired nucleotides (e.g.
0and1). The shadow describes the secondary structure in less detail than the bracket notation.The list of pairing indices is a sequence of integers that each indicate to which nucleotide a nucleotide in the sequence is paired. For instance, in the zero-based list
[2, -1, 0], the first nucleotide is paired to the third nucleotide. The value-1designates an unpaired nucleotide. CT files use one-based lists of pairing indices. It can represent pseudo-knots.The scoring matrix presents a list of pairing indices in a 2D matrix. It can represent pseudo-knots.
Alternatively, this problem can be described as “multiple binary classification problems that predict whether each pair of nucleotides forms a base pair or not” [5].
RNA secondary structure prediction methods usually comprise the following stages:
Pre-processing transforms the primary structure (i.e. sequence of letters) into another representation.
Prediction converts the input into an output using a previously trained model.
Post-processing transforms the output of the model into a secondary structure (e.g. bracket notation or shadow).
Definitions
Term |
Definition |
|---|---|
contact matrix or pairing scoring matrix |
A 2D matrix in which 0 indicates a non-paired base and 1 indicates a paired base. Can represent a secondary structure with pseudo-knots [1] [7]. |
Classical Prediction Models
The term “classical model” is used in this context to refer to predictive models that do not rely on neural network.
MXFold2
RNAfold
RNAstructure
TORNADO
Prediction Models Based on Neural Networks
This section summarizes a few recent RNA secondary structure prediction models.
REDfold (2023)
REDfold [9] uses a contact matrix as the input and an encoder-decoder for the processing. It supports pseudoknots.
CNNFold (2022)
CNNFold [1] is a predictive model designed by Booy, Ilin, and Orponen that uses no energy model and can predict pseudo-knots.
The input of the model is a matrix of possible pairings. Each cell indicates if the pairing is possible and, if so, what bases would be paired to one another, as shown in the figure below:
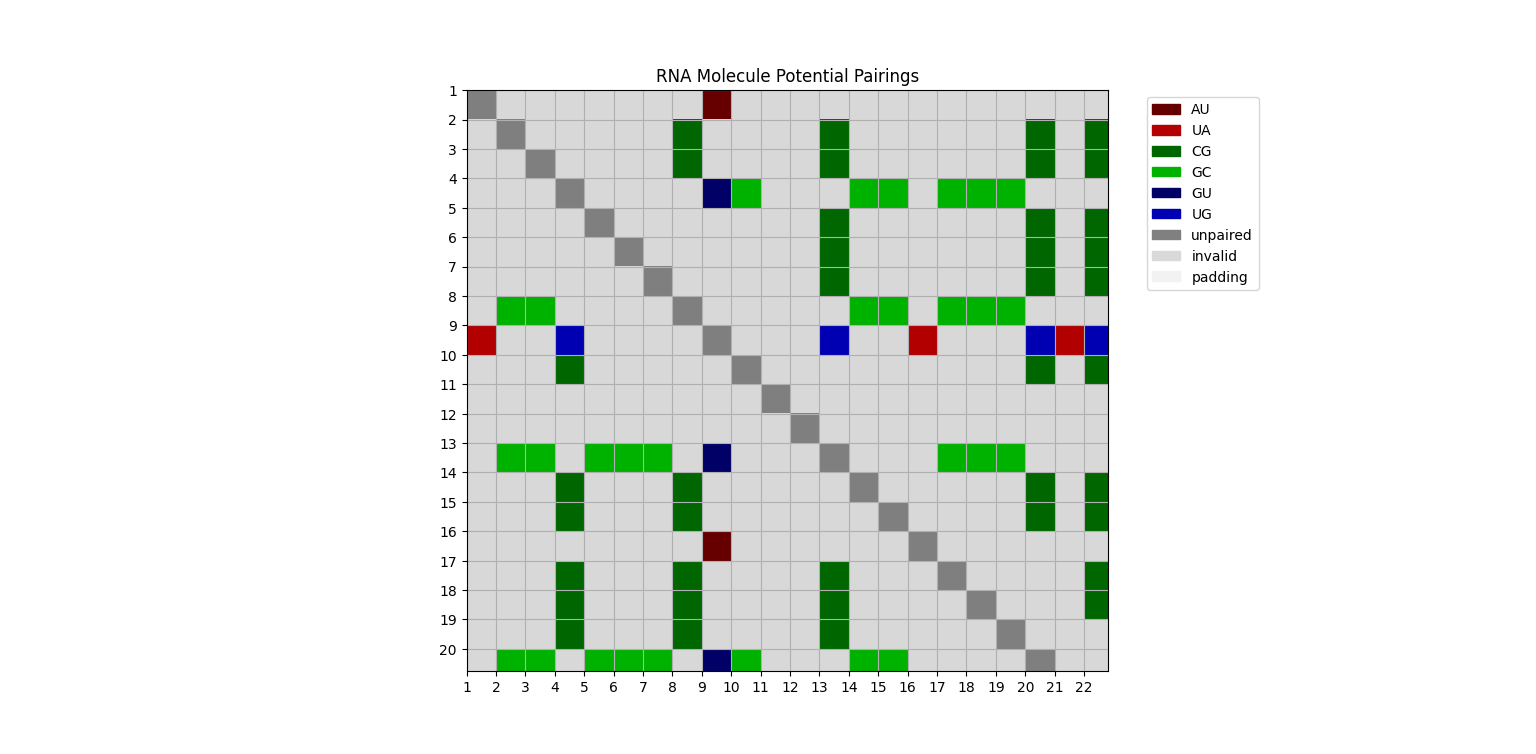
Example of a matrix of potential pairings.
This representation makes the predictive model more efficient because it captures global information about the structure of the molecule. Instead of only using the local information of a primary structure (i.e. a nucleotide and its neighboring nucleotides), the model can rely on the pre-processed potential pairings.
The predictive model is a deep convolutional neural network. It uses only the pairing matrix as the input and does not consider any energy model.
The output is a scoring matrix that is converted to a secondary structure.
ATTFold (2020)
ATTFold [6] is a predictive model designed by Wang, Liu, Gao, Zhang, and Dong that uses no energy model and can predict pseudo-knots.
The input is a one-hot encoded primary structure.
The model is a pipeline of components:
The first component is a transformer network that encodes the primary structure.
A CNN follows the transformer and decodes the output of the transformer into a base pairing scoring matrix.
Hard constraints are then applied to the scoring matrix to (1) remove impossible pairings (e.g.
AtoC) and (2) remove pairings less than three bases apart.
The output is a base pairing scoring matrix.
UFold (2022)
UFold [7] is a predictive model designed by Fu, Cao, Wu, Peng, Nie, and Xie that uses no energy model and can predict pseudo-knots.
The input is a contact matrix, which is generated by computing the Kronecker product of the one-hot encoded primary structure with itself. This data structure differs from the matrix of possible pairings used in CNNFold.
The model is a U-Net neural network.
The output is a base pairing scoring matrix.
MXFold2 (2021)
MXFold2 [5] is a predictive model designed by Sato, Akiyama, and Sakakibara that uses an energy model and cannot predict pseudo-knots.
The input is the one-hot encoded primary structure.
The model is a pipeline of components:
A BiLSTM recurrent neural network.
Convolutional and concatenation layers.
A Zucker-style dynamic programming algorithm (THAT’s the energy model, right there).
The output is the secondary structure in dot-bracket notation.
CDPFold (2019)
CDPFold [8] is a predictive model designed by Zhang, Li, and Wei that uses no energy model and cannot predict pseudo-knots.
The input of the model is a matrix-encoded encoded primary structure whose element represent possible base pairings. This is similar to CNNFold, but the elements of the matrix are scalars instead of vectors.
The model is a pipeline of components:
A CNN predicts base pairing probabilities.
A maximum probability sum algorithm (a modified Nussimov dynamic programming method) corrects predictions to obtain valid secondary structures.
The output is the secondary structure in dot-bracket notation.
SPOT-RNA (2019)
SPOT-RNA [10] is a predictive model designed by Singh, Hanson, Paliwal, and Zhou that uses no energy model and can predict pseudo-knots.
The model uses ensemble and transfer learning to predict structures.
General Observations
Szikszai et al. [11] show that most models based on neural networks perform poorly at family-wise cross validation. In other words, they have a hard time generalizing predictions to unfamiliar structures.
Datasets
[4]
RNAStralign: https://www.urmc.rochester.edu/rna/
RNAalign: https://drive.google.com/drive/folders/19KPRYJjjMJh1qdMhtmUoYA_ncw3ocAHc
RFam: https://rfam.org/
References
- 1(1,2)
Mehdi Saman Booy, Alexander Ilin, and Pekka Orponen. Rna secondary structure prediction with convolutional neural networks. BMC Bioinformatics, 23(1):58, 2022. URL: https://doi.org/10.1186/s12859-021-04540-7, doi:10.1186/s12859-021-04540-7.
- 2
Accessed: 2023-07-15. URL: http://rna.tbi.univie.ac.at/forna/.
- 3
David Mathews. Mathews lab. Accessed: 2023-04-15. URL: https://rna.urmc.rochester.edu/.
- 4
Andronescu, Bereg, Hoos, and Condon. Rna strand v2.0 - the rna secondary structure and statistical analysis database. 2009. Accessed: 2023-04-15. URL: http://www.rnasoft.ca/strand/.
- 5(1,2)
Kengo Sato, Manato Akiyama, and Yasubumi Sakakibara. Rna secondary structure prediction using deep learning with thermodynamic integration. Nature Communications, 12(1):941, 2021. URL: https://doi.org/10.1038/s41467-021-21194-4, doi:10.1038/s41467-021-21194-4.
- 6
Yili Wang, Yuanning Liu, Shuo Wang, Zhen Liu, Yubing Gao, Hao Zhang, and Liyan Dong. Attfold: rna secondary structure prediction with pseudoknots based on attention mechanism. Frontiers in Genetics, 2020. URL: https://www.frontiersin.org/articles/10.3389/fgene.2020.612086, doi:10.3389/fgene.2020.612086.
- 7(1,2)
Laiyi Fu, Yingxin Cao, Jie Wu, Qinke Peng, Qing Nie, and Xiaohui Xie. UFold: fast and accurate RNA secondary structure prediction with deep learning. Nucleic Acids Research, 50(3):e14–e14, 11 2021. URL: https://doi.org/10.1093/nar/gkab1074, arXiv:https://academic.oup.com/nar/article-pdf/50/3/e14/42544496/gkab1074\_supplemental\_file.pdf, doi:10.1093/nar/gkab1074.
- 8
Hui Zhang, Cong Zhang, Zhe Li, Chunhua Li, Xiaopeng Wei, Baofeng Zhang, and Yunlong Liu. A new method of rna secondary structure prediction based on convolutional neural network and dynamic programming. Frontiers in Genetics, 10:467, 2019. doi:10.3389/fgene.2019.00467.
- 9
missing author in redfold
- 10
Jaswinder Singh, Jack Hanson, Kuldip Paliwal, and Yaoqi Zhou. Rna secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nature Communications, 10:2041–1723, 2019. doi:10.1038/s41467-019-13395-9.
- 11
Marcell Szikszai, Michael Wise, Amitava Datta, Max Ward, and David H Mathews. Deep learning models for RNA secondary structure prediction (probably) do not generalize across families. Bioinformatics, 38(16):3892–3899, 06 2022. URL: https://doi.org/10.1093/bioinformatics/btac415, arXiv:https://academic.oup.com/bioinformatics/article-pdf/38/16/3892/45300927/btac415\_supplementary\_data.pdf, doi:10.1093/bioinformatics/btac415.